

Una malattia rara, senza una cura concreta, viene rappresentata in un film del 2019 diretto da Justin Baldoni.

Non sempre si è a conoscenza dei nuovi flagelli che colpiscono la nostra specie ed è per questo che il film ‘’A un metro da te’’ è stato ideato e prodotto.
‘’ A un metro da te ‘’: l’amore contro la fibrosi cistica
Stella Grant è affetta da fibrosi cistica, una patologia molto grave che compromette polmoni, pancreas ed intestino. Nonostante ciò, la ragazza decide di condividere la sua patologia nel mondo dei social network, raccontando delle sue giornate tutte uguali a cercare di sconfiggere un mostro a dir poco immortale. Ha un compagno di avventure, Poe, anche lui affetto dalla sua stessa malattia, con il quale condivide dolori e sofferenze che, ormai, le restano accanto da una vita. Un giorno, però, conosce Will Newman, anche lui affetto da fibrosi cistica, che si sta sottoponendo a delle cure sperimentali per combattere una grave infezione batterica polmonare, conseguenza della patologia che entrambi condividono. I due si avvicinano sempre di più, si aprono e raccontano l’uno all’altro le loro giornate, i loro pensieri e i loro sogni, sperando in un futuro senza quella patologia che tanto li ha uniti. I due si innamorano, ma non possono avvicinarsi fisicamente l’uno all’altro, per evitare le cosiddette ‘’infezioni crociate’’ che due persone affette da fibrosi cistica possono trasmettersi. Ci sono, dunque, dei limiti fisici che impediscono ai due ragazzi di vivere come due normali adolescenti alle prese con la prima storia d’amore, devono stare ad almeno due metri di distanza e non possono nemmeno sfiorarsi con una carezza. Will spera che la cura sperimentale porti a degli esisti positivi, limitando almeno le infezioni polmonari, ma i dottori ritengono che non ci sia una soluzione concreta che possa portare il ragazzo ad un vera guarigione. Il destino di Will è ormai segnato, la morte è più vicino di quello che sembra e dovrà dire addio a Stella.

La fibrosi cistica: una malattia genetica rara
La fibrosi cistica è una malattia molto grave, congenita, dovuta ad una mutazione del gene CFTR. La malattia è autosomica recessiva, quindi colui che nasce affetto ha due copie del gene malato ereditate da due genitori normalmente portatori sani della malattia ma inconsapevoli del gene mutato. Il gene sano codifica per una proteina chiamata CFTR ovvero Cystic Fibrosis Transmembrane Conductance Regulator deputata a regolare il corretto funzionamento della secrezione di molti organi, come il pancreas e intestino. Chi ha la fibrosi cistica possiede una proteina non funzionante che altera tutti i meccanismi di scambi ionici che quotidianamente avvengono nelle cellule di trachea, pancreas e intestino. Le secrezioni dei pazienti affetti risultano più dense, disidratate e poco fluide che danneggiano l’organo secernente. Gli organi più colpiti sono i bronchi e i polmoni, i quali secernono muco per eliminare tramite la faringe batteri e sostanze nocive intrappolate nelle vie aeree. Se le secrezioni sono troppo dense, la clearance mucociliare tracheale viene alterata poichè le cellule ciliate dell’epitelio respiratorio non riescono a spingere in faringe il muco con le sostanze nocive intrappolate. Questo porta ad un ristagno di batteri e patogeni che sviluppa gravi infezioni delle vie aeree. A livello del pancreas, gli enzimi non riescono ed essere secreti nell’intestino poichè il succo pancreatico è troppo denso e questo porta ad alterazioni della digestione, malassorbimento, diarrea e ritardo nella crescita. Il progredire del danno pancreatico porta ad una forma di diabete. Nell’intestino, fegato, cavità nasali e dotti deferenti del maschio possono manifestarsi complicanze anche molto gravi. Anche le ghiandole sudoripare risentono della proteina mutata producendo sudore troppo salato, evento che rende il cosiddetto ‘’test del sudore’’ fondamentale per la diagnosi di fibrosi cistica.
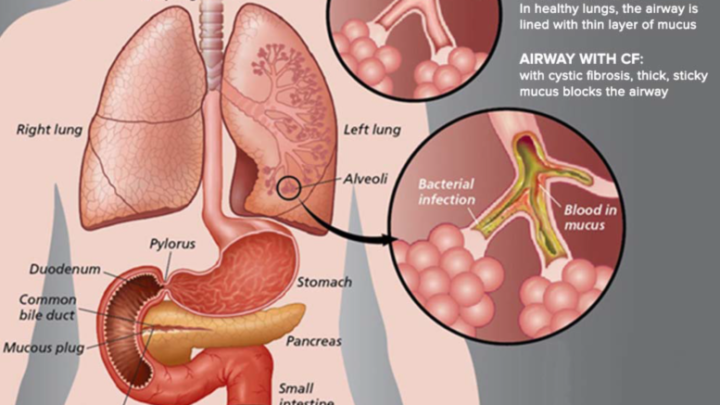
Il destino dei pazienti affetti da fibrosi cistica
Putroppo, ad oggi, le cure sono dirette alla cura dei sintomi e delle complicanze, senza andare a risolvere il problema di fondo, ovvero al mutazione genetica congenita. Ci sono protocolli internazionali che vengono adattati al singolo malato. In generale si somministrano antibiotici per le infezioni polmonari, aerosol di antibiotici, farmaci fluidificanti per le secrezioni troppo dense, fisioterapia respiratoria, enzimi digestivi per sopperire alla carenza dovuta alla secrezione pancreatica eccessivamente viscosa, dieta ipercalorica e trattamento delle complicanze eventuali. Il trapianto polmonare è obbligatorio per i pazienti con insufficienza respiratoria irreversibile. Bisogno anche considerare che i sintomi e la gravità di questa patologia così complessa variano da soggetto a soggetto ed è per questo che ogni paziente ha un suo iter farmacologico e riabilitativo. Il gene CFTR può avere diverse tipologie di mutazioni che portano a diverse varianti di fibrosi cistica, contribuendo a rendere questa patologia ancora più complessa e variabile. La ricerca sta lavorando a nuove cure sperimentali per questa malattia, arrivando a garantire una sopravvivenza molto più elevata rispetto al secolo scorso, quando i bambini affetti da questa malattia non arrivavano nemmeno ai 5 o 6 anni di età. Si sono scoperti farmaci che riescono ad intervenire su date tipologie di mutazione genetica. Oggi ci sono molti adulti affetti da questa malattia che lavorano, studiano, hanno dei figli e convivono con questo flagello combattendo giorno dopo giorno, anche se di certo al qualità della loro vita è piuttosto scarsa. Si ritiene che, i malati, non riescano ad arrivare ad un età media superiore ai 40 anni, a causa delle svariate complicazioni correlate alla loro mutazione genetica.
