

I vampiri sono delle figure mitologiche notturne, che cercano persone da cui poter succhiare sangue come nutrimento. Ma esistono patologie che rendono l’uomo simile a un vampiro?

La figura del vampiro, già nota nel folclore del 1700, è stata ripetutamente immortalata in opere cinematografiche come Twilight o in libri come Dracula, il capolavoro di Bram Stoker.
Dracula: Bram Stoker e il conte Vlad
Nel 1897, l’autore irlandese Stoker pubblica un romanzo che riscuoterà un enorme successo nei secoli successivi, incentrato sulla figura del Conte Dracula, personaggio che poi ha ispirato molti tratti del moderno concetto di vampirismo.
Il racconto viene descritto mediante delle lettere, articoli di giornare e sono tutte disposte in ordine cronologico, esponendo i fatti dal punto di vista di vari personaggi. Il conte Dracula, nel racconto, cerca di fuggire dalla Transilvania e raggiungere l’Inghilterra dove potrebbe trovare nuove vittime e diffondere il vampirismo. Tuttavia, l’antagonista (buono?) della vicenda è Van Helsing, un professore e dottore noto internazionalmente come cacciatore di vampiri.
Tornando alla realtà, fuori dal romanzo, ha affascinato da sempre dopo la pubblicazione del racconto di Stoker la figura del leggendario Conte Vlad III, noto anche come Vlad Dracula, figlio di Vlad Dracul che era stato imperatore della Wallachia nel 1436. A causa della sua crudeltà (veniva anche detto impalatore perchè fece impalare i due inviati del Sultano Ottomano Mehmed II venuti in visita per rendere omaggio), alla base di aneddoti che iniziarono a diffondersi durante il suo periodo di prigionia fino al 1475 in Ungheria, Stoker si ispirò al suo nome per trarre il suo personaggio principale, il conte Dracula.

Emoglobina ed eme: le basi del trasporto di ossigeno
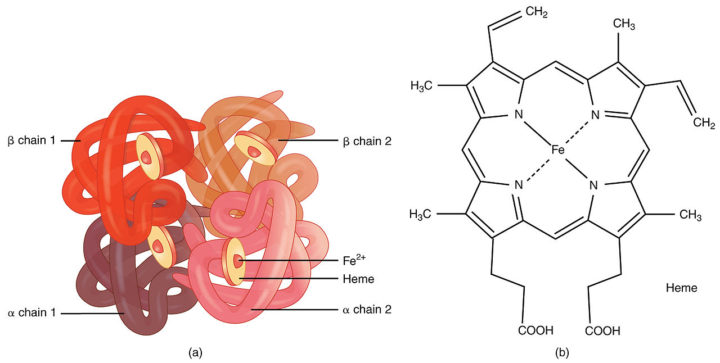
L’ossigeno molecolare viene trasportato in circolo nel sangue mediante l’emoglobina, un tetramero di proteine globulari caratterizzate ciascuna dalla presenza centrale di un gruppo eme, composto da un anello tetrapirrolico noto come porfirina e da uno ione di ferro centrale coordinato con quattro atomi di N dell’anello precedentemente citato. L’emoglobina presenta solitamente due stati, T, quando essa non lega l’ossigeno (viene anche detta deossiemoglobina e viene stabilizzata da alcuni composti come gli idrogenioni o l’anidride carbonica, secondo l’effetto Bohr), e R, quando invece lega l’ossigeno al ferro, “caricando” l’emoglobina del prezioso gas che viene successivamente portato in circolo nel sangue fino ai tessuti che ne hanno necessità.
La biosintesi dell’eme è un processo estremamente complicato, che parte dal succinil-CoA e dalla glicina, sostanze unite fra loro da un enzima noto come ALAS, ossia acido aminolevulinico sintasi di cui esistono due isoforme, la ALAS-1 presente in quasi tutti i tessuti e la ALAS-2, che invece risulta essere specifica degli eritrociti. Due molecole di acido aminolevulinico sono fuse fra loro generando il porfibilinogeno, da cui poi si avvia un processo di sintesi multi-step che si conclude con la protoporfirina IX. Quest’ultima subisce l’effetto della ferrochelatasi, enzima che aggiunge il ferro centrale portando alla formazione dell’eme finale, nello specifico dell’eme b.
Porfirie: quando la biosintesi dell’eme non va a buon fine
Un qualsiasi difetto genico a carico di uno degli enzimi coinvolti nella biosintesi delle porfirine può determinare un accumulo metabolico pericoloso di sostanze intermedie. Nei casi più lievi, si può verificare un accumulo di acido aminolevulinico, di coproporfirinogeno IX o di protoporfirina IX che possono essere espulsi dal nostro organismo mediante le vie cataboliche o di biotrasformazione. Si ricorda che un accumulo di protoporfirina può anche essere dovuto ad una carenza di ferro, aggiunto dalla ferrochelatasi, tipico delle anemie ferroprive.
Nei casi più gravi possiamo avere della malattie come:
- Porfiria acuta intermittente = causata da mutazioni della porfibilinogeno deidrogenasi, si trasmette in maniera autosomica dominante e causa fortissimi dolori addominali. Si possono verificare anche seri danni neurologici;
- Porfiria congenit eritropoietica = dovuta a mutazioni di uroporfibilinogeno III sintasi, con conseguente accumulo metabolico di idrossimetilbilano. Si tratta di una malattia autosomica recessiva che fa accumulare invece uroporfibilinogeno I che si forma spontaneamente. Solitamente si manifesta con acuta fotosensibilità per cui gli individui devono assolutamente evitare l’esposizione alla luce solare;
- Porfiria cutanea tarda = si tratta della forma più diffusa di porfiria, caratterizzata da accumulo di uroporfibilinogeno III. Determina fotosensibilità, iperpigmentazione (probabilmente adattativa) e ipertricosi. Solitamente la malattia può essere secondaria, come avviene con determinati problemi epatici o può insorgere da un substrato genetico nel qual caso si tratta di una malattia autosomica dominante;
