Dei ricercatori hanno pubblicato sul The New England Journal of Medicine due nuovi protocolli sperimentali per curare due malattie genetiche molto comuni.
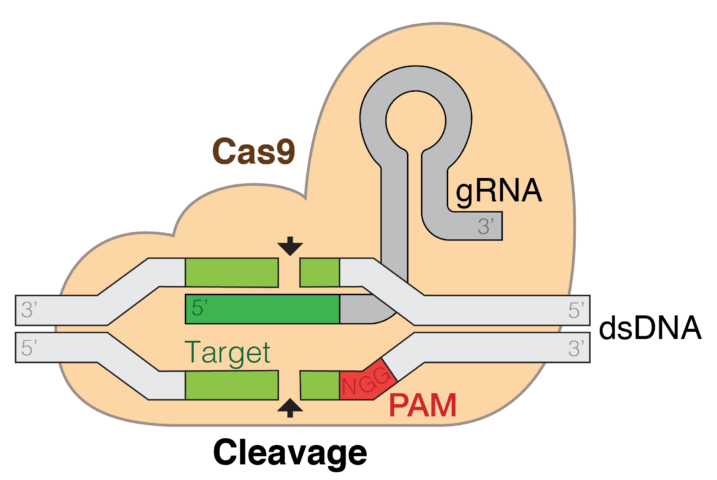
I due protocolli, di cui uno si basa sulla tecnologia CRISPR, permettono di risolvere e trattare i pazienti con una terapia genetica contro la beta talassemia e la anemia falciforme.
La scoperta dei ricercatori: la terapia genica
I nuovi trattamenti proposti da questo team sono promettenti ma sembrano necessitare ancora di molto follow-up, siccome si tratta di interventi che richiedono sia delicati prelievi dal midollo osseo dei pazienti, sia risultano essere molto costosi. Nei due nuovi trattamenti, gli investigatori hanno rimosso le cellule staminali ematiche del paziente e, nel laboratorio, hanno disattivato un interruttore genetico detto BCL11A che, nella vita iniziale, spegne i geni per la forma fetale dell’emoglobina. I pazienti ricevono poi una chemioterapia per fare piazza pulita delle cellule danneggiate e queste cellule staminali modificate in laboratorio vengono nuovamente trapiantate nel paziente. Col gene della emoglobina fetale attiva, le proteine fetali riescono a ripristinare l’emoglobina nel contesto della talassemia. Invece, nell’anemia falciforme, permette di rimpiazzare alcune cellule a forma di falce, bloccando anche la formazione dei tactoidi polimerizzanti. Gli scienziati hanno ingegnerizzato l’enzima CRISPR per tagliare il gene BCL11A e, in uno sforzo genetico più tradizionale, un altro team del Boston Children’s Hospital, ha raggiunto lo stesso risultato. Infatti, loro hanno utilizzato un virus reso inoffensivo per iniettare nel genoma delle cellule staminali ematiche, un pezzo di DNA codificante per un RNA che silenzia l’interruttore inibente la sintesi dell’emoglobina fetale.
Anemia falciforme: cause e caratteristiche
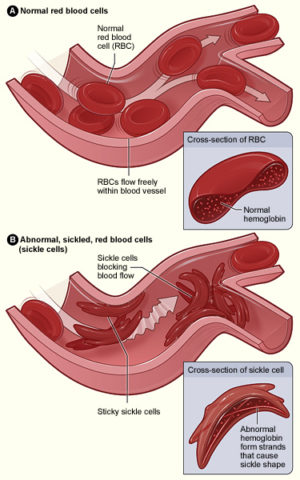
Si parla di anemia falciforme riferendosi ad un gruppo di patologie, specificamente una condizione ereditaria in cui non ci sono abbastanza globuli rossi sani da poter portare ossigeno attraverso il corpo umano. La patologia spesso assume un pattern autosomico recessivo e si associa a mutazioni puntiforme nel gene codificante per la globina B. Infatti, questa variante di emoglobina, detta anche HbS, è dovuta ad un SNP (single nucleotide polymorphism) associato ad una sostituzione dell’acido glutammico con una valina. Solitamente, le cellule rosse si muovono facilmente nei vasi sanguigni mentre nell’anemia falciforme assumono la forma di luna crescente: questo conferisce una rigidità strutturale che comporta poi un accumulo e blocco delle cellule stesse nei piccoli vasi sanguigni. Il sintomo più comune e anche caratteristico è appunto la anemia, ossia una rottura delle cellule e la loro morte precoce (10-20 giorni al posto dei caratteristici 120). I pazienti potrebbero anche sperimentare una maggiore suscettibilità alle infezioni, dovuta a ripetuti danneggiamenti del microcircolo della milza, coinvolta anche nella protezione dalle infezioni. Spesso infatti i medici prescrivono vaccinazioni o antibiotici a bambini affetti dalla patologia per evitare che insorgano delle sovrainfezioni, come la pneumonia. Talvolta si potrebbero anche osservare danni visivi, siccome si ha ostruzione dei vasi ematici che riforniscono la retina, ossia la porzione iniziale della via ottica encefalica, rivestente i 2/3 posteriori della camera posteriore dell’occhio.
Beta-talassemia: causa e caratteristiche
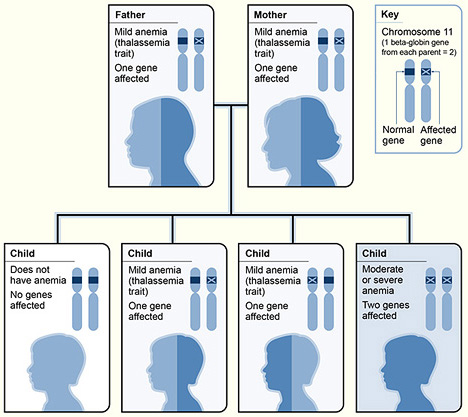
Si tratta di un gruppo di disordini ematologici ereditari, caratterizzati da una ridotta o assente sintesi delle catene beta della emoglobina, con sintomatologia variabile in base alla gravità della condizione. Alla base di questa patologia possiamo trovare sia delle forme da non-delezione (difetti che in generale sono basati su un SNP o una piccola inserzione prima del gene della beta globina; talvolta questo si osserva nei promoter, determinando una riduzione della sintesi della catena globinica) sia nelle forme da delezione (si possono verificare delle patologie molto diversificate da un punto di vista sintomatologico, come la HPFT, ossia la persistenza ereditaria dell’emoglobina fetale). In base all’accoppiamento dei due alleli per il gene della beta globina, i pazienti possono essere diagnosticati con la talassemia minor (anemia microcitica con lieve riduzione del valore del volume corpuscolare medio degli eritrociti), la talassemia intermedia (gli individui affetti possono anche vivere una vita normale ma è probabile che debbano sottoporsi a trasfusioni in periodi particolari come malattie croniche o gravidanze) e la talassemia maior, per cui si ha una severa anemia microcitica ipocromica che potrebbe causare sia splenomegalia (ingrandimento della milza) che enormi deformità alla struttura dell’osso e del relativo midollo.