

La fibrosi cistica è la malattia ereditaria mortale più comune in Europa. Ripercorriamo la trama del film “A un metro da te” per capire di cosa si tratta.

La storia d’amore che nasce in ospedale tra Stella Grant e Will Newman è ostacolata dal motivo per cui si trovano lì, la fibrosi cistica. Quest’ultima è una della malattie più comuni nell’uomo, è una malattia ereditaria autosomica recessiva causata da una mutazione sul gene CFTR.
La divulgazione di Stella
Stella è una ragazza affetta da fibrosi cistica, terminale alla nascita, e ha necessariamente bisogno di un trapianto di polmoni. Stella condivide le sue giornate postando video su YouTube in cui parla della sua malattia senza mai perdere il sorriso. Si trova in ospedale, quando un giorno conosce un ragazzo, di nome Will, anch’egli malato di CF. L’infermiera le raccomanda di tenersi lontano dal ragazzo per evitare il rischio di infezioni crociate, ovvero di infezioni batteriche tra i pazienti, che, nella loro situazione, sarebbero potute essere fatali. Will si trova in ospedale per una terapia sperimentale, che non molto più tardi si rivela inefficace, nel frattempo i due si innamorano e si supportano a vicenda restando ad una distanza di sicurezza. La fibrosi cistica è la malattia ereditaria mortale più comune, si manifesta con la stessa frequenza nei maschi e nelle femmine ed è una della malattie che più sono in grado di accorciare la vita. Negli Stati Uniti sono circa 30 mila le persone affette, in Europa 1 persona su 25 è portatrice di una delle mutazioni che possono causare la malattia, sono infatti conosciute circa 1600 mutazioni che possono causare la fibrosi cistica, alcune a carico dalla sequenza genica di CFTR altre (circa il 13%) a carico dello splicing del gene, più avanti sarà tutto più chiaro.
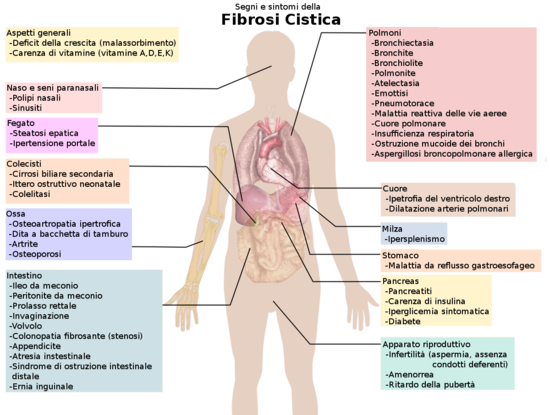
Fibrosi Cistica, segni e sintomi
Di cosa soffrono i due amanti? Cerchiamo di capire la generalità della malattia. I sintomi della fibrosi cistica coinvolgono disfunzioni degli organi interni causati dalla mancanza o poca presenza di una proteina codificata dal gene CFTR, ma di questo parleremo nel dettaglio dopo. Chi soffre di fibrosi cistica presenta un largo raggio di sintomi, riguardanti polmoni, pancreas, intestino, milza, cuore, ossa, fegato, apparato riproduttivo, colecisti. Difficile pensare che tutto questo possa essere causato dall’assenza di una sola proteina. La proteina in questione, però, si trova sulla membrana delle cellule epiteliali ed è addetta al trasporto di ioni cloro delle cellule degli organi o apparati appena menzionati. Questo “piccolo” dettaglio si traduce in un enorme squilibrio cellulare che a sua volta si traduce in sintomi gravi. Abbiamo innanzitutto un deficit della crescita causata dal malassorbimento dei nutrienti, quindi le persone affette pur seguendo una dieta bilanciata presentano scarsa crescita e scarso aumento di peso, e per lo stesso concetto, una carenza vitaminica. Per quanto riguarda i polmoni, invece, questa anomalia provoca una secrezione di muco denso e viscoso che a sua volta causa infezioni polmonari ricorrenti. Il problema “ostruzione” riguarda anche i problemi intestinali, come protrusioni interne e incapacità di escrezione delle feci, inoltre, i liquidi densi del pancreas bloccano il movimento degli enzimi digestivi nel duodeno e provocano danni irreversibili al pancreas, spesso sfociando in una dolorosa infiammazione, e la mancanza di enzimi digestivi porta a difficoltà di assorbire i nutrienti, con la loro successiva escrezione nelle feci portando al malassorbimento di cui abbiamo parlato prima. Inoltre danni a carico del pancreas possono portare a squilibri dell’insulina causando una forma di diabete tipica della malattia. Inoltre la questione malassorbimento ritorna quando parliamo dei sintomi alle ossa, infatti la carenza di vitamina D, coinvolta nella regolazione di calcio e fosfato, può portare all’osteoporosi. Inoltre, le persone affette sono solite prendere antibiotici a causa dell’elevato rischio di infezioni che potrebbero portare a polmoniti acute. Entriamo un po’ più nei dettagli molecolari di una variante della fibrosi cistica.
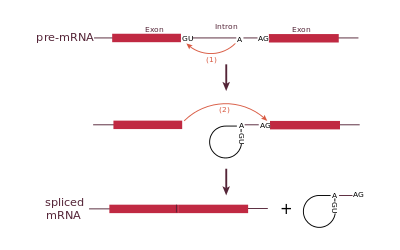
Una variante della CF
La variante di cui parliamo riguarda la produzione di uno splicing aberrante. Nel nostro DNA sono presenti zone codificanti che portano alla produzione di proteine, lavoro svolto dalle RNA polimerasi. Una volta ottenuto un trascritto di queste zone, esse devono essere “maturate” prima di poter diventare delle proteine a tutti gli effetti. Una delle tappe riguardanti la maturazione è un processo chiamato splicing, in cui sono rimosse delle zone non codificanti chiamati introni, insomma, il trascritto è accorciato e, successivamente, tradotto in proteina dai ribosomi. Cosa c’entra questo con la fibrosi cistica? Il gene di cui abbiamo parlato, CFTR, essendo un gene codificante proteina è soggetto a splicing. Alcune mutazioni portano all’accorciamento di una sequenza degli introni, quelli che devono essere rimossi, utile proprio per discriminare le porzioni codificanti (che devono rimanere) da quelle non codificanti (introni). L’accorciamento di questa sequenza provoca un non riconoscimento del confine tra introni ed esoni (porzione codificante) e per questo l’esone 9 viene eliminato insieme all’introne presente e successivo. Questo provoca una sequenza che pur tradotta non porterebbe alla formazione della proteina utile al trasporto del cloro, con le dovute conseguenze di cui abbiamo parlato precedentemente.
